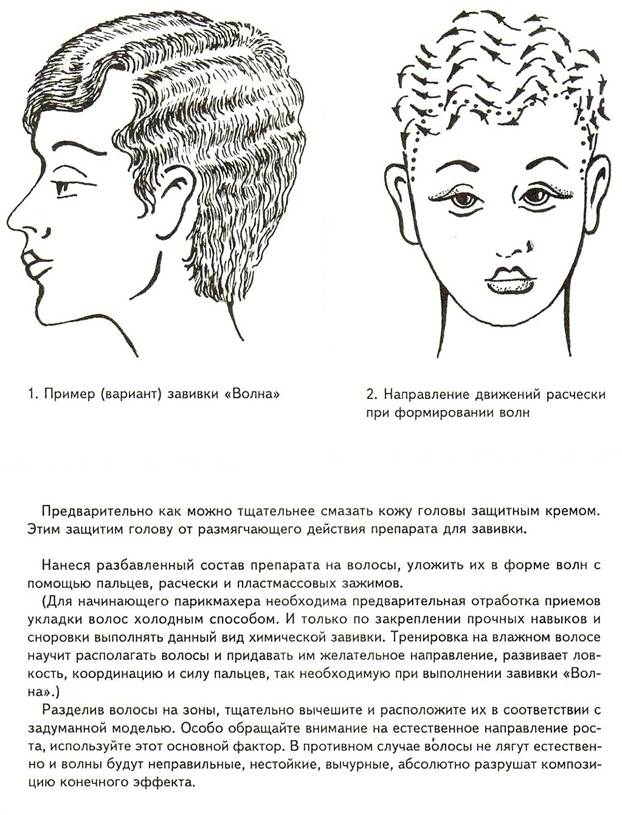
Всё о стрижках и прическах: Последовательность выполнения химической завивки
Материалы и инструменты. В процессе химической завивки волос используют такие материалы, инструменты и принадлежности:
1) средство для выполнения завивки;
2) средство для фиксации;
3) шампунь;
4) бальзам для возобновления структуры волос;
5) коклюшки определенного диаметра;
6) гребень с «хвостиком»;
7) миски для препаратов, достаточно стойкие, во избежание случайного переворачивания во время работы;
8) защитные рукавицы;
9) губки, спонжи для нанесения препаратов;
10) зажимы для волос;
11) отеплительный колпак;
12) полотенца.
Подготовительные работы. К подготовительным работам перед химической завивкой относятся:
1) внешний|наружный| обзор|осмотр| кожи головы и волос;
2) отбор химического препарата по типу волос;
3) проверка кожи головы на раздражение препаратом;
4) проверка состояния|стана| волос при смачивании препаратом;
5) выбор вида завивки волос;
6) выбор метода нанесения препарата на волосы;
7) выбор метода и размера коклюшек;
8) мойка головы;
9) стрижка волос.
Внешний|наружный| обзор|осмотр| кожи головы и волос необходим для выявления| разнообразных|многообразных| заболеваний кожи и волос, при которых|каких| химическую завивку выполнять|исполнять| запрещено|запретный|. Противопоказанием для выполнения| химической завивки волос является наличие на коже царапин, порезов, язв и других ярко выраженных раздражений.
Важно оценить|оценивать| состояние|стан| волос, чтобы сделать|совершить| правильный выбор препарата для химзавивки|. Необходимо определить структуру волос (тонкая, толстая, нормальная), качество (мягкое, среднее, жесткое), вид предыдущей|предварительной| обработки (крашенный|крашеное|, осветлённый|осветленное|, с остатками перманента).
После выбора смеси следует проверить реакцию кожи головы клиента на действие препарата. Для этого нужно кусочком ваты, смоченным| в препарате, нанести смесь на кожу| за ушной раковиной. Через|из-за| 8-10 мин| будет заметная реакция кожи на раздражение. При отсутствии на коже покраснения считают, что смесь клиенту не противопоказана|. Если будет обнаружено|выявляет| раздражение, нанесенную смесь нужно смыть водой, а от химической завивки этим препаратом отказаться.|
Если будет обнаружено|выявляет| раздражение, нанесенную смесь нужно смыть водой, а от химической завивки этим препаратом отказаться.|
Одновременно с проверкой реакции кожи на раздражение необходимо проверить реакцию волос на действие на него препарата для завивки.
При осмотре|осмотре| волос следует обратить внимание на его состояние|стан|. В случае, когда волосы поддавались|подвергают| сильному обесцвечению или окрашиванию|окраске| химическими| красителями, нужно проверить его на разрыв. Если волосы в сухом состоянии|стане| легко разрываются, завивку выполнять|исполнять| запрещено|запретный|.
Для пробы химпрепарат| наносят на тонкую прядь|пасмо| волос, через|из-за| 3-5 мин| испытывают волосы на разрыв. Если волосы сохраняют|берегут| прочность — препарат подходит для завивки, если же они легко разрываются|, необходимо подобрать смесь более слабой концентрации.
Технологии секвенирования и анализы: где мы были и куда мы идем?
- Список журналов
- iScience
- т.
 18; 2019 авг 30
18; 2019 авг 30 - PMC6733309
 18; 2019 авг 30
18; 2019 авг 30Являясь библиотекой, NLM предоставляет доступ к научной литературе. Включение в базу данных NLM не означает одобрения или согласия с содержание NLM или Национальных институтов здравоохранения. Узнайте больше о нашем отказе от ответственности.
iScience. 2019 30 августа; 18: 37–41.
Опубликовано в Интернете 15 августа 2019 г. doi: 10.1016/j.isci.2019.06.035
1 и
Информация об авторе Информация об авторских правах и лицензии Отказ от ответственности 9001 3
Волна технологий более десяти лет назад превратила секвенирование в эра высокой пропускной способности, требующая исследований новых вычислительных методов для анализа этих данных. С тех пор области применения этих технологий секвенирования постоянно расширялись. Семинар RECOMB Satellite Workshop on Massively Parallel Sequencing (RECOMB-Seq), учрежденный в 2011 году, объединяет ведущих исследователей в области вычислительной геномики и геномной биологии для обсуждения новых возможностей в разработке алгоритмов для массивно-параллельного секвенирования данных. Девятый выпуск этого семинара был проведен в Вашингтоне, округ Колумбия, в Университете Джорджа Вашингтона 3 и 4 мая 2019 г.. Было изучено несколько традиционных тем анализа последовательностей, включая сборку генома, выравнивание последовательностей и сжатие данных, а также разработка методов для новых технологий секвенирования, включая связанные чтения и секвенирование с длительным чтением одной молекулы. Здесь мы возвращаемся к этим темам и обсуждаем текущее состояние и перспективы технологий секвенирования и анализов.
Семинар RECOMB Satellite Workshop on Massively Parallel Sequencing (RECOMB-Seq), учрежденный в 2011 году, объединяет ведущих исследователей в области вычислительной геномики и геномной биологии для обсуждения новых возможностей в разработке алгоритмов для массивно-параллельного секвенирования данных. Девятый выпуск этого семинара был проведен в Вашингтоне, округ Колумбия, в Университете Джорджа Вашингтона 3 и 4 мая 2019 г.. Было изучено несколько традиционных тем анализа последовательностей, включая сборку генома, выравнивание последовательностей и сжатие данных, а также разработка методов для новых технологий секвенирования, включая связанные чтения и секвенирование с длительным чтением одной молекулы. Здесь мы возвращаемся к этим темам и обсуждаем текущее состояние и перспективы технологий секвенирования и анализов.
Достижения в высокопроизводительных технологиях секвенирования обеспечивают целостные исследовательские возможности для решения критически важных и сложных проблем практически во всех областях биологии. Эти технологии привели к взрывному росту объема данных секвенирования, генерируемых каждый год. Например, проект «Геном человека» стоил миллиарды долларов, и на его реализацию ушло десятилетие, тогда как за последние 5 лет было секвенировано более 100 000 геномов человека. Помимо повышения производительности и стоимости, появился ряд новых технологий секвенирования, в том числе секвенирование со сверхдлинным считыванием (например, Nanopore), рестрикционные карты высокого разрешения (например, данные Bionano), технологии секвенирования со связанным считыванием и методы перекрестного связывания (см. обзор различных технологий секвенирования). Все эти технологии секвенирования были источником дискуссий и интенсивных исследований. Здесь мы суммируем некоторые из самых последних открытий и возможностей.
Эти технологии привели к взрывному росту объема данных секвенирования, генерируемых каждый год. Например, проект «Геном человека» стоил миллиарды долларов, и на его реализацию ушло десятилетие, тогда как за последние 5 лет было секвенировано более 100 000 геномов человека. Помимо повышения производительности и стоимости, появился ряд новых технологий секвенирования, в том числе секвенирование со сверхдлинным считыванием (например, Nanopore), рестрикционные карты высокого разрешения (например, данные Bionano), технологии секвенирования со связанным считыванием и методы перекрестного связывания (см. обзор различных технологий секвенирования). Все эти технологии секвенирования были источником дискуссий и интенсивных исследований. Здесь мы суммируем некоторые из самых последних открытий и возможностей.
Таблица 1
Сравнение длины считывания, частоты ошибок и стоимости различных технологий секвенирования ДНК
| Технология | Метод | Длина считывания | 9 0038 Частота ошибок (%) Пропускная способность (ГБ/ пробег) | |
|---|---|---|---|---|
| Illumina | Синтез | 100–300 п. н. н. | 0,1 | 200–600 |
| Pacific Biosciences SMRT | Синтез | 10–100 kb | 5–15 | 10–20 |
| Oxford Nanopore MinION | Nanopore | Переменная (до 1000 кб) | 5–20 | 5–10 |
Открыть в отдельном окне
Сборка геномов с помощью дробового секвенирования хромосомной ДНК по-прежнему остается фундаментальной проблемой в сообществе биоинформатики. Как заявил Адам Филлиппи в своем выступлении «40 лет сборки генома: мы еще не закончили?» разработка стратегий сборки прочтений последовательностей была описана в конце 19 в.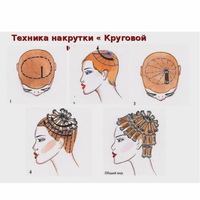

Для преодоления этих проблем был предложен ряд вычислительных решений, которые улучшают качество как сборки, так и данных последовательности. Морисс и соавт. (2019) предложил метод самоисправления длинных чтений, который сочетает в себе алгоритмические подходы современных современных методов исправления ошибок длинных чтений, а именно построение и использование множественного выравнивания чтений, а впоследствии , граф де Брейна. Они демонстрируют, что этот метод способен исправлять ошибки как при чтении длинных, так и сверхдлинных последовательностей, и обладает высокой масштабируемостью, поскольку это единственный метод, который можно масштабировать до набора данных человека, содержащего сверхдлинные чтения. Марийон и др. (2019) описывают метод анализа графов сборки, полученных в результате длинных чтений, для восстановления контигов, потерянных в процессе сборки. Они демонстрируют, что их метод восстанавливает полезную информацию о смежности между контигами, и показывают, что он способен «обеспечивать более информативное представление фрагментированных сборок, исследовать повторяющиеся структуры и предлагать вероятные порядки контигов».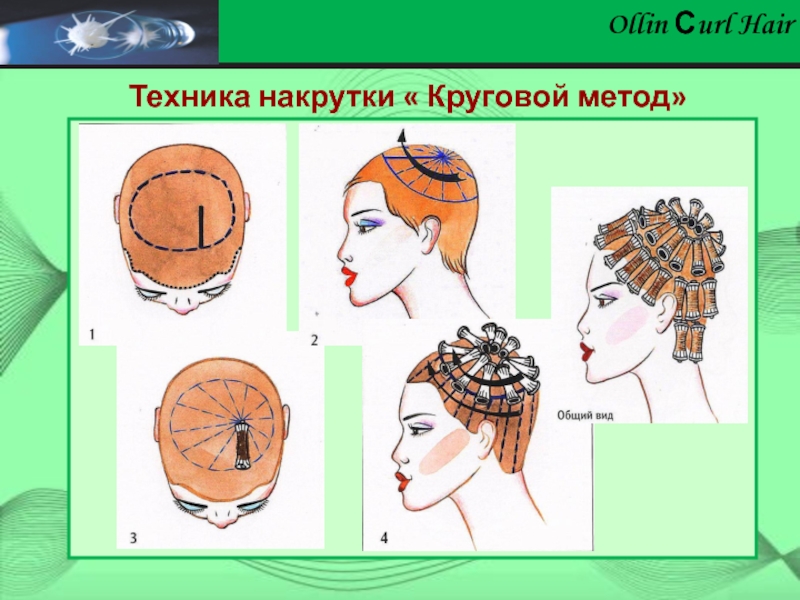
Для видов с собранным геномом прочтенные последовательности могут быть сопоставлены с этим эталонным геномом для идентификации генетических вариантов и выполнения множества других биологических анализов. Следовательно, выравнивание последовательностей ДНК с геномом является фундаментальной вычислительной проблемой. За последние 10 лет был разработан ряд методов для решения проблемы сопоставления коротких прочтений (длиной 50–200 оснований) с эталонным геномом (Reinert et al., 2015). Многие из этих инструментов выравнивания были разработаны специально для выравнивания прочтений, полученных с использованием протоколов секвенирования следующего поколения (NGS), таких как секвенирование РНК (RNA-seq) и секвенирование микроРНК (miRNA). Чтения, созданные с использованием RNA-seq, могут охватывать соединения экзон-экзон, и поэтому для точного картирования чтений RNA-seq требуется способность обнаруживать сплайсированные выравнивания. Чжун и Чжан (2019)) описали инструмент выравнивания, предназначенный для обеспечения точного картирования перекрестно-сшитых прочтений микроРНК-мРНК. Этот инструмент использует индекс на основе преобразования Берроуза-Уилера (BWT) для поиска коротких совпадений, но реализует ряд дополнительных оптимизаций, позволяющих обеспечить точное картирование дуплексных прочтений, образованных взаимодействиями микроРНК-мРНК.
Следовательно, выравнивание последовательностей ДНК с геномом является фундаментальной вычислительной проблемой. За последние 10 лет был разработан ряд методов для решения проблемы сопоставления коротких прочтений (длиной 50–200 оснований) с эталонным геномом (Reinert et al., 2015). Многие из этих инструментов выравнивания были разработаны специально для выравнивания прочтений, полученных с использованием протоколов секвенирования следующего поколения (NGS), таких как секвенирование РНК (RNA-seq) и секвенирование микроРНК (miRNA). Чтения, созданные с использованием RNA-seq, могут охватывать соединения экзон-экзон, и поэтому для точного картирования чтений RNA-seq требуется способность обнаруживать сплайсированные выравнивания. Чжун и Чжан (2019)) описали инструмент выравнивания, предназначенный для обеспечения точного картирования перекрестно-сшитых прочтений микроРНК-мРНК. Этот инструмент использует индекс на основе преобразования Берроуза-Уилера (BWT) для поиска коротких совпадений, но реализует ряд дополнительных оптимизаций, позволяющих обеспечить точное картирование дуплексных прочтений, образованных взаимодействиями микроРНК-мРНК.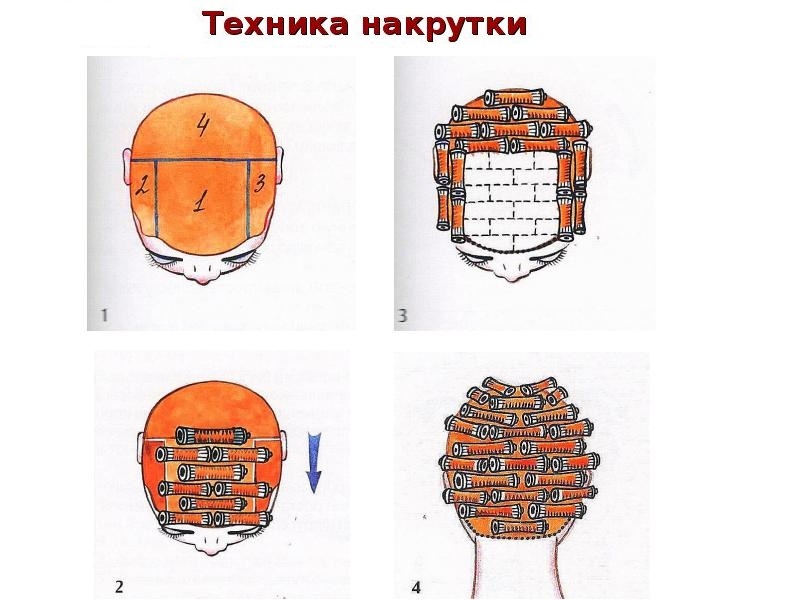
С появлением технологий секвенирования длинных считываний одиночных молекул, таких как Pacific Biosciences SMRT и Oxford Nanopore MinION (Pollard et al., 2018), возрастает потребность в инструментах выравнивания, способных выравнивать длинные считывания. Существующие инструменты выравнивания NGS оптимизированы для низкого уровня ошибок и короткой длины считывания, тогда как эти технологии генерируют считывания длиной в десятки килобаз и с высоким уровнем ошибок (5–20 %, см. Ресурсы). Почти все инструменты выравнивания с коротким чтением используют хеш-таблицу или индекс на основе BWT для эффективного поиска коротких совпадений между последовательностью запроса и геномом. Подходы, основанные на хеш-таблицах, требуют хранения большого индекса для поиска исходных совпадений, что может быть недопустимым для больших геномов, таких как геномы человека.
Обнаружение генетических вариантов с использованием чтения последовательности, выровненной с эталонным геномом, возможно, является наиболее распространенным применением технологий NGS. Подобно выравниванию прочтений, было разработано множество инструментов для обнаружения вариантов коротких последовательностей (однонуклеотидных вариантов и коротких вставок или делеций). Используя современные инструменты, такие как GATK (DePristo et al., 2011), оба эти типа вариантов могут быть надежно обнаружены с помощью секвенирования ДНК всего генома или всего экзома. Тем не менее, другие типы вариантов, такие как структурные варианты, по-прежнему сложно обнаружить с помощью чтения NGS. Мелисса Гимрек выделила одно такое ограничение NGS для коротких тандемных повторов (STR). STR (тандемные повторы мотивов длиной от 1 до 6 оснований) широко распространены в геноме человека и склонны к мутациям, которые могут расширять или сокращать повтор. Было показано, что экспансии STR вызывают ряд редких менделевских заболеваний (Ashley, 2016). Одним из примеров такого заболевания является болезнь Хантингтона, которая вызывается экспансией тринуклеотидного повтора. Генотипирование STR и обнаружение повторяющихся экспансий требует тщательного анализа для захвата сигнала для таких событий в чтении коротких последовательностей. Гимрек описал вычислительный инструмент GangSTR (Mousavi et al., 2019), которые могут точно генотипировать STR в более чем 500 000 локусов тандемных повторов и даже обнаруживать экспансии повторов, которые длиннее, чем длина ридов Illumina. Тем не менее, в этой области остается много проблем, включая генотипирование GC-богатых повторов и учет неоднородности охвата последовательностей.
Тем не менее, другие типы вариантов, такие как структурные варианты, по-прежнему сложно обнаружить с помощью чтения NGS. Мелисса Гимрек выделила одно такое ограничение NGS для коротких тандемных повторов (STR). STR (тандемные повторы мотивов длиной от 1 до 6 оснований) широко распространены в геноме человека и склонны к мутациям, которые могут расширять или сокращать повтор. Было показано, что экспансии STR вызывают ряд редких менделевских заболеваний (Ashley, 2016). Одним из примеров такого заболевания является болезнь Хантингтона, которая вызывается экспансией тринуклеотидного повтора. Генотипирование STR и обнаружение повторяющихся экспансий требует тщательного анализа для захвата сигнала для таких событий в чтении коротких последовательностей. Гимрек описал вычислительный инструмент GangSTR (Mousavi et al., 2019), которые могут точно генотипировать STR в более чем 500 000 локусов тандемных повторов и даже обнаруживать экспансии повторов, которые длиннее, чем длина ридов Illumina. Тем не менее, в этой области остается много проблем, включая генотипирование GC-богатых повторов и учет неоднородности охвата последовательностей.
Подобно картированию считывания, обнаружение вариантов в различных приложениях (например, соматических вариантов в геномах рака) требует специализированных инструментов. Шарлотта Дарби представила умный подход (Дарби и др., 2019 г.) для обнаружения вариантов мозаики с использованием технологии связанного чтения 10X Genomics (Zheng et al., 2016). В отличие от вариантов зародышевой линии мозаичные варианты — это те, которые присутствуют только в подмножестве клеток человека и их труднее обнаружить. В отличие от стандартного секвенирования Illumina, связанные чтения предоставляют информацию о гаплотипах дальнего действия, которую можно использовать для различения мозаичных мутаций (присутствующих в подмножестве прочтений одного гаплотипа) от ошибок секвенирования и других артефактов. Новый метод, Самовар, присваивает чтения гаплотипам с использованием связанных прочтений и позволяет точно обнаруживать мозаичные мутации в геномах детского рака без использования сопоставления нормальных наборов данных.
Такие инструменты, как GangSTR и Samovar, имеют решающее значение для реализации всего потенциала полногеномного секвенирования и будут способствовать дальнейшему расширению использования NGS в качестве диагностического инструмента. Одним из ограничений этих инструментов является то, что они основаны на выравнивании прочтений с эталонным геномом и предназначены для обнаружения конкретных типов вариантов. Растет интерес к методам обнаружения вариантов и генотипирования без выравнивания для данных NGS. Такие методы используют информацию, содержащуюся в наборе k-меров (и их количестве), наблюдаемых в считываниях последовательностей, и могут использоваться для обнаружения почти всех типов вариантов последовательностей (Nordstrom et al., 2013). Даниэль Стэндидж представил метод, основанный на кевларе (Standage et al., 2019 г.).), который выявляет 90 109 вариантов de novo 90 110 в геноме человека путем выявления частых k-меров, которые либо полностью отсутствуют, либо очень часто появляются в геномах родителей. Этот подход без выравнивания может обнаруживать SNV, короткие вставки и даже структурные варианты. Подходы без выравнивания также ценны для генотипирования известных вариантов в секвенированном геноме. Лука Денти описал MALVA (Bernardini et al., 2019), который может эффективно генотипировать как SNV, так и короткие вставки и улучшает предыдущие методы решения этой проблемы. Успех методов без выравнивания предполагает, что подходы, сочетающие анализ на основе k-mer с эталонным картированием, могут максимизировать точность обнаружения вариантов и генотипирования с использованием чтения NGS.
Этот подход без выравнивания может обнаруживать SNV, короткие вставки и даже структурные варианты. Подходы без выравнивания также ценны для генотипирования известных вариантов в секвенированном геноме. Лука Денти описал MALVA (Bernardini et al., 2019), который может эффективно генотипировать как SNV, так и короткие вставки и улучшает предыдущие методы решения этой проблемы. Успех методов без выравнивания предполагает, что подходы, сочетающие анализ на основе k-mer с эталонным картированием, могут максимизировать точность обнаружения вариантов и генотипирования с использованием чтения NGS.
Проект 1000 геномов (Консорциум проекта 1000 геномов, 2015 г.) в настоящее время в основном завершен, и сейчас проект 100 000 геномов идет полным ходом (Turnbull et al., 2018). Без сжатия необработанные данные для 100 000 геномов человека требуют примерно 300 терабайт дискового пространства. Учитывая размер данных и их постоянный рост, эффективное сжатие и распаковка данных жизненно важны для любого вида анализа. Существуют различные методы сжатия данных, которые часто, но не обязательно зависят от целей анализа. Общие алгоритмы сжатия, такие как анализ Лемпеля-Зива (Зив и Лемпель, 1977), BWT (Burrows and Wheeler, 1994) и кодирование Хаффмана (Turnbull et al., 2018) направлены на преобразование входного файла (файлов) в представление, требующее меньшего количества битов, чем исходный файл (файлы). И наоборот, распаковка направлена на восстановление исходных файлов из сжатого формата.
Существуют различные методы сжатия данных, которые часто, но не обязательно зависят от целей анализа. Общие алгоритмы сжатия, такие как анализ Лемпеля-Зива (Зив и Лемпель, 1977), BWT (Burrows and Wheeler, 1994) и кодирование Хаффмана (Turnbull et al., 2018) направлены на преобразование входного файла (файлов) в представление, требующее меньшего количества битов, чем исходный файл (файлы). И наоборот, распаковка направлена на восстановление исходных файлов из сжатого формата.
Данные последовательности предлагают уникальные возможности для значительного сжатия, поскольку они содержат высокий уровень избыточности. Был разработан ряд методов, использующих новые структуры данных для использования этой характеристики для достижения эффективного сжатия и распаковки. Деревья последовательности Блума и компактные представления графа де Брейна — два примера таких структур данных, которые постоянно улучшались в последние несколько лет. Sequence Bloom Trees были впервые предложены Соломоном и Кингсфордом (2016) в качестве средства для эффективного индексирования данных о последовательности таким образом, чтобы поддерживать запросы о наличии транскриптов. SeqOthello (Yu et al., 2018), Split-SBT (Solomon and Kingsford, 2018) и AllSome-SBT (Sun et al., 2017) улучшают это недавнее исходное представление. Павел Медведев описал представление (Harris and Medvedev, 2019), который требует значительно меньше времени и места для построения индекса, демонстрируя, что остается возможность для дальнейшего улучшения существующих представлений. Для сравнения, графы де Брейна, которые первоначально были предложены для сборки генома, использовались для компактного индексирования всех тыс. -меров длины ( тыс. -меров) из набора прочтений последовательностей. Несмотря на многочисленные улучшения в представлении графов де Брейна (Muggli et al., 2017, Almodaresi et al., 2019, Karasikov et al., 2019, Almodaresi et al., 2017, Alipanahi et al., 2018, Mustafa et al., 2017, Pandey et al., 2018), мы по-прежнему наблюдаем существенные улучшения существующих представлений. Например, представление Marchet et al. (2019) удалось проиндексировать все тыс.
SeqOthello (Yu et al., 2018), Split-SBT (Solomon and Kingsford, 2018) и AllSome-SBT (Sun et al., 2017) улучшают это недавнее исходное представление. Павел Медведев описал представление (Harris and Medvedev, 2019), который требует значительно меньше времени и места для построения индекса, демонстрируя, что остается возможность для дальнейшего улучшения существующих представлений. Для сравнения, графы де Брейна, которые первоначально были предложены для сборки генома, использовались для компактного индексирования всех тыс. -меров длины ( тыс. -меров) из набора прочтений последовательностей. Несмотря на многочисленные улучшения в представлении графов де Брейна (Muggli et al., 2017, Almodaresi et al., 2019, Karasikov et al., 2019, Almodaresi et al., 2017, Alipanahi et al., 2018, Mustafa et al., 2017, Pandey et al., 2018), мы по-прежнему наблюдаем существенные улучшения существующих представлений. Например, представление Marchet et al. (2019) удалось проиндексировать все тыс. -меров из генома человека в пространстве 8 ГБ и 30 минут и все тыс. -меров из -меров аксолотля (в 10 раз больше человеческого генома) за 63 -GB пространство и в течение 10 ч. Эта область использования графов де Брейна для компактного представления и индексации k -mers еще имеет неизведанные направления.
-меров из генома человека в пространстве 8 ГБ и 30 минут и все тыс. -меров из -меров аксолотля (в 10 раз больше человеческого генома) за 63 -GB пространство и в течение 10 ч. Эта область использования графов де Брейна для компактного представления и индексации k -mers еще имеет неизведанные направления.
Наконец, предстоит еще большая работа по разработке целевого параллелизма для быстрого сжатия и распаковки больших файлов gzip. Кербириу и Райан представили параллельный алгоритм быстрой распаковки файлов, сжатых gzip, который обеспечивает произвольный доступ к сжатым данным о последовательности ДНК в формате FASTQ. Демонстрация их метода показывает, что он на порядок быстрее, чем gunzip, и в пять раз быстрее, чем высокооптимизированная последовательная реализация.
Две повторяющиеся темы были затронуты на RECOMB-Seq 2019. Во-первых, хотя вычислительные методы выравнивания, сборки и обнаружения вариантов значительно продвинулись за последнее десятилетие, остаются серьезные проблемы, например, сквозная сборка генома с использованием длинных чтений и обнаружение повторяющихся вариантов с помощью высокопроизводительного секвенирования. Во-вторых, необходимы новые алгоритмы и структуры данных для обработки данных, полученных из нескольких геномов, с использованием новейших технологий секвенирования. В частности, технологии секвенирования с длительным чтением, такие как Pacific Biosciences SMRT и секвенирование со связанным чтением 10X Genomics, становятся все более распространенными, и мы ожидаем увидеть разработку новых методов, использующих эти технологии в ближайшем будущем. Наконец, встреча не увенчалась бы успехом без усердной работы членов программного и руководящего комитетов. Мы хотели бы поблагодарить всех, кто внес свой вклад в создание RECOMB-Seq 2019.успех.
Во-вторых, необходимы новые алгоритмы и структуры данных для обработки данных, полученных из нескольких геномов, с использованием новейших технологий секвенирования. В частности, технологии секвенирования с длительным чтением, такие как Pacific Biosciences SMRT и секвенирование со связанным чтением 10X Genomics, становятся все более распространенными, и мы ожидаем увидеть разработку новых методов, использующих эти технологии в ближайшем будущем. Наконец, встреча не увенчалась бы успехом без усердной работы членов программного и руководящего комитетов. Мы хотели бы поблагодарить всех, кто внес свой вклад в создание RECOMB-Seq 2019.успех.
CB был профинансирован NIH через грант NIAID R01AI141810-01 и NSF через грант IIS-1618814.
- Алипанахи Б., Кунле А. и Буше К. (2018). Перекрашивание раскрашенного графа де Брейна. В: Proceedings of String Processing and Information Retrieval (SPIRE 2018), стр. 1–11.
- Альмодареси Ф., Панди П. и Патро Р. (2017). Rainbowfish: краткое цветное графическое представление де Брейна. В: Материалы семинара по алгоритмам в биоинформатике (WABI 2017), стр. 251–265.
- Альмодарези Ф., Пандей П., Фердман М., Джонсон Р. и Патро Р. (2019). Эффективное, масштабируемое и точное представление многомерной информации о цвете благодаря поиску по графу де Брейна. В: Материалы исследований в области вычислительной молекулярной биологии (RECOMB 2019), стр. 1–18.
- Эшли Э.А. На пути к прецизионной медицине. Нац. Преподобный Жене. 2016;17:507–522. [PubMed] [Google Scholar]
- Бернардини Г., Бониццони П., Денти Л., Превитали М., Александр Шёнхут А. МАЛЬВА: генотипирование путем обнаружения аллелей известных вариантов без картирования. БиоРксив. 2019[Бесплатная статья PMC] [PubMed] [Google Scholar]
- Burrows, M. and Wheeler, D.J. (1994). Алгоритм сжатия данных без потерь с сортировкой блоков, Технический отчет 124, Digital Equipment Corporation, https://www.hpl.hp.com/techreports/Compaq-DEC/SRC-RR-124.pdf.
- Дарби К.А., Фитч Дж.Р., Бреннан П. Дж., Келли Б.Дж., Бир Н., Магрини В., Леонард Дж., Коттрелл К.Е., Гастье-Фостер Дж.М., Уилсон Р.К. Самовар: одновыборочный мозаичный вызов SNV со связанными чтениями. iНаука. 2019 [Бесплатная статья PMC] [PubMed] [Google Scholar]
- ДеПристо М.А., Кинтеро Дж.К., Круз Д.Ф., Кинтеро К., Хабманн Г., Фульки-Морено М.Р., Верстрепен К.Дж., Тевелейн Дж.М., Томе Дж. Структура для обнаружения вариаций и генотипирования с использованием данных секвенирования ДНК следующего поколения. Нац. Жене. 2011;43:491–498. [Бесплатная статья PMC] [PubMed] [Google Scholar]
- Харрис Р.С., Медведев П. Улучшено представление последовательности деревьев Блума. БиоРксив. 2019 г.: 10.1101/501452. [CrossRef] [Google Scholar]
- Карасиков М., Мустафа Х., Джоудаки А., Джавадзаде-Но С., Рач Г. и Калес А. (2019). Разреженные представления бинарных отношений для аннотации графа генома. В: Материалы исследований в области вычислительной молекулярной биологии (RECOMB 2019). стр. 120–135. 10.1101/468512. [CrossRef]
- Li H. Minimap2: попарное выравнивание нуклеотидных последовательностей. Биоинформатика. 2018;34:3094–3100. [Бесплатная статья PMC] [PubMed] [Google Scholar]
- Marchet C., Kerbiriou M., Limasset A. Индексирование графиков де Брейна с помощью минимизаторов. БиоРксив. 2019 г.: 10.1101/546309. [Перекрестная ссылка] [Академия Google]
- Марийон П., Чихи Р., Варре Ж.С. Графический анализ фрагментированных сборок бактериального генома с длительным чтением. Биоинформатика. 2019 btz219, https://academic.oup.com/bioinformatics/advance-article-abstract/doi/10.1093/bioinformatics/btz219/5421164?redirectedFrom=fulltext. [PubMed] [Google Scholar]
- Морисс П., Марше К., Лимассет А., Лекрок Т., Лефевр А. СОГЛАСИЕ: масштабируемая самокоррекция длинных ридов с множественным выравниванием последовательностей. БиоРксив. 2019 г.: 10.1101/546630. [Перекрестная ссылка] [Академия Google]
- Мусави Н., Шлейзер-Бурко С., Яницкий Р., Гымрек М. Профилирование полногеномного ландшафта тандемных повторов. Нуклеиновые Кислоты Res. 2019 [бесплатная статья PMC] [PubMed] [Google Scholar]
- Muggli MD, Bowe A., Noyes NR, Morley P.S., Belk KE, Raymond R., Gagie T., Puglisi SJ, Boucher C. Краткие цветные графики де Брейна . Биоинформатика. 2017; 33:3181–3187. [Бесплатная статья PMC] [PubMed] [Google Scholar]
- Мустафа Х., Калес А., Карасиков М., Раетч Г. Метаннот: краткая структура данных для сжатия цветов в динамических графах де Брейна. БиоРксив. 2017 год: 10.3929/ethz-b-000236153. [CrossRef] [Google Scholar]
- Nordstrom K.J., Albani M.C., James G.V., Gutjahr C., Hartwig B., Turck F., Paszkowski U., Coupland G., Schneeberger K. Идентификация мутаций путем прямого сравнения всего генома данные секвенирования мутантов и особей дикого типа с использованием k-меров. Нац. Биотехнолог. 2013;31:325–330. [PubMed] [Google Scholar]
- Пандей П., Альмодареси Ф., Бендер М.А., Фердман М., Джонсон Р., Патро Р. Мантис: быстрый, небольшой и точный крупномасштабный индекс поиска последовательности. Клетка. 2018;7:201–207. [PubMed] [Академия Google]
- Поллард М.О., Гурдасани Д., Ментцер А.Дж., Портер Т., Сандху М.С. Лонгриды: их назначение и место. Гум. Мол. Жене. 2018;27(R2):R234–R241. [Бесплатная статья PMC] [PubMed] [Google Scholar]
- Reinert K., Langmead B., Weese D., Evers D.J. Выравнивание показаний секвенирования следующего поколения. Анну. Преподобный Геномикс Хам. Жене. 2015;16:133–151. [PubMed] [Google Scholar]
- Робертс М., Хейс В., Хант Б.Р., Маунт С.М., Йорк Дж.А. Снижение требований к хранению для сравнения биологических последовательностей. Биоинформатика. 2004; 20:3363–3369. [PubMed] [Google Scholar]
- Шлемов А., Коробейников А. PathRacer: гоночный профиль трасс HMM на графе сборки. БиоРксив. 2019 г.: 10.1101/562579. [CrossRef] [Google Scholar]
- Соломон Б., Кингсфорд К. Быстрый поиск среди тысяч экспериментов по секвенированию с коротким считыванием. Нац. Биотехнолог. 2016 [бесплатная статья PMC] [PubMed] [Google Scholar]
- Соломон Б. , Кингсфорд К. Улучшенный поиск в больших базах данных транскриптомного секвенирования с использованием деревьев расщепленной последовательности. Дж. Вычисл. биол. 2018;25:755–765. [Бесплатная статья PMC] [PubMed] [Google Scholar]
- Стаден Р. Стратегия секвенирования ДНК с использованием компьютерных программ. Нуклеиновые Кислоты Res. 1979; 6: 2601–2610. [Бесплатная статья PMC] [PubMed] [Google Scholar]
- Standage DS, Titus Brown C., Hormozdiari F. Kevlar: не картографическая структура для точного обнаружения вариантов de novo. БиоРксив. 2019 [Бесплатная статья PMC] [PubMed] [Google Scholar]
- Сун, К., Харрис, Р.С., Чихи, Р., и Медведев П. (2017). AllSome последовательность цветения деревьев. В: Proceedings of the Research in Computational Molecular Biology (RECOMB 2017), стр. 272–286, https://doi.org/10.1007/9.78-3-319-56970-3_17.
- Консорциум проекта «1000 геномов» Глобальный справочник по генетическим вариациям человека. Природа. 2015; 526: 68–74. [Бесплатная статья PMC] [PubMed] [Google Scholar]
- Turnbull C. , Scott R.H., Thomas E., Jones L., Murugaesu N., Pretty F.B., Halai D., Baple E., Craig C., Hamblin A , Проект 100 000 геномов: секвенирование всего генома в NHS. бр. Мед. Дж. 2018;361:k1687. [PubMed] [Google Scholar]
- Yu Y., Liu J., Liu X., Zhang Y., Magner E., Lehnert E., Qian C., Liu J. SeqOthello: опрос экспериментов по секвенированию РНК в масштабе. Геном биол. 2018;19:167. [Бесплатная статья PMC] [PubMed] [Google Scholar]
- Zheng GX, Lau BT, Schnall-Levin M., Jarosz M., Bell JM, Hindson C.M., Kyriazopoulou-Panagiotopoulou S., Masquelier DA, Merrill L., Terry Дж. М. Гаплотипирование зародышевых и раковых геномов с помощью высокопроизводительного секвенирования связанного чтения. Нац. Биотехнолог. 2016; 34:303–311. [Бесплатная статья PMC] [PubMed] [Google Scholar]
- Zhong C., Zhang S. CLAN: CrossLinked читает инструмент ANalyais. iНаука. 2016 [Google Scholar]
- Зив Дж., Лемпель А. Универсальный алгоритм последовательного сжатия данных. IEEE Trans. Инф. Теория. 1977;23:337–343. [Google Scholar]
 В: Материалы семинара по алгоритмам в биоинформатике (WABI 2017), стр. 251–265.
В: Материалы семинара по алгоритмам в биоинформатике (WABI 2017), стр. 251–265. Дж., Келли Б.Дж., Бир Н., Магрини В., Леонард Дж., Коттрелл К.Е., Гастье-Фостер Дж.М., Уилсон Р.К. Самовар: одновыборочный мозаичный вызов SNV со связанными чтениями. iНаука. 2019 [Бесплатная статья PMC] [PubMed] [Google Scholar]
Дж., Келли Б.Дж., Бир Н., Магрини В., Леонард Дж., Коттрелл К.Е., Гастье-Фостер Дж.М., Уилсон Р.К. Самовар: одновыборочный мозаичный вызов SNV со связанными чтениями. iНаука. 2019 [Бесплатная статья PMC] [PubMed] [Google Scholar]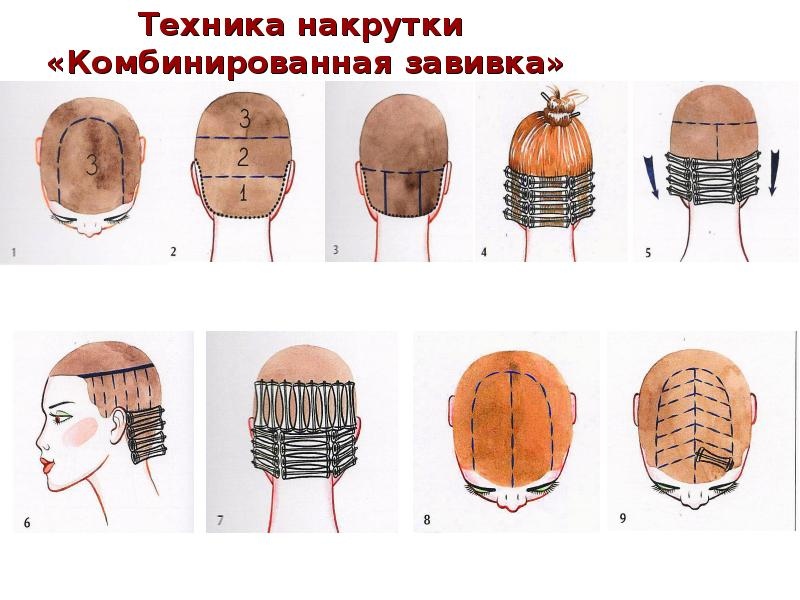 [CrossRef]
[CrossRef] Нуклеиновые Кислоты Res. 2019 [бесплатная статья PMC] [PubMed] [Google Scholar]
Нуклеиновые Кислоты Res. 2019 [бесплатная статья PMC] [PubMed] [Google Scholar] Клетка. 2018;7:201–207. [PubMed] [Академия Google]
Клетка. 2018;7:201–207. [PubMed] [Академия Google] , Кингсфорд К. Улучшенный поиск в больших базах данных транскриптомного секвенирования с использованием деревьев расщепленной последовательности. Дж. Вычисл. биол. 2018;25:755–765. [Бесплатная статья PMC] [PubMed] [Google Scholar]
, Кингсфорд К. Улучшенный поиск в больших базах данных транскриптомного секвенирования с использованием деревьев расщепленной последовательности. Дж. Вычисл. биол. 2018;25:755–765. [Бесплатная статья PMC] [PubMed] [Google Scholar] , Scott R.H., Thomas E., Jones L., Murugaesu N., Pretty F.B., Halai D., Baple E., Craig C., Hamblin A , Проект 100 000 геномов: секвенирование всего генома в NHS. бр. Мед. Дж. 2018;361:k1687. [PubMed] [Google Scholar]
, Scott R.H., Thomas E., Jones L., Murugaesu N., Pretty F.B., Halai D., Baple E., Craig C., Hamblin A , Проект 100 000 геномов: секвенирование всего генома в NHS. бр. Мед. Дж. 2018;361:k1687. [PubMed] [Google Scholar] Инф. Теория. 1977;23:337–343. [Google Scholar]
Инф. Теория. 1977;23:337–343. [Google Scholar]Статьи из iScience предоставлены здесь с разрешения Elsevier
Секвенирование нового поколения (NGS) | Знакомство с технологией
Секвенирование нового поколения для начинающих
Мы познакомим вас с основами NGS, предоставив учебные пособия и советы по планированию вашего первого эксперимента.
Начало работыЧто такое NGS?
Секвенирование нового поколения (NGS) — это технология массового параллельного секвенирования, обеспечивающая сверхвысокую производительность, масштабируемость и скорость. Эта технология используется для определения порядка нуклеотидов во всех геномах или целевых областях ДНК или РНК. NGS произвела революцию в биологических науках, позволив лабораториям выполнять широкий спектр приложений и изучать биологические системы на невиданном ранее уровне.
Современные сложные вопросы геномики требуют такой глубины информации, которая недоступна традиционным технологиям секвенирования ДНК. NGS заполнил этот пробел и стал повседневным инструментом для решения этих вопросов.
NGS заполнил этот пробел и стал повседневным инструментом для решения этих вопросов.
Применение NGS
Технология секвенирования следующего поколения коренным образом изменила виды вопросов, которые ученые могут задавать и на которые могут ответить. Инновационные возможности пробоподготовки и анализа данных обеспечивают широкий спектр применений. Например, NGS позволяет лабораториям:
- Быстрое секвенирование полных геномов
- Глубокое секвенирование целевых областей
- Использование секвенирования РНК (RNA-Seq) для обнаружения новых вариантов РНК и сайтов сплайсинга или количественного определения мРНК для анализа экспрессии генов
- Анализ эпигенетических факторов, таких как полногеномное метилирование ДНК и взаимодействие ДНК-белок
- Секвенирование образцов рака для изучения редких соматических вариантов, субклонов опухоли и многого другого
- Исследование микробиома человека
- Выявление новых патогенов
Изучение методов и способов секвенирования
 youtube.com/embed/fCd6B5HRaZ8?rel=0&showinfo=0&enablejsapi=1″ frameborder=»0″>
youtube.com/embed/fCd6B5HRaZ8?rel=0&showinfo=0&enablejsapi=1″ frameborder=»0″> Как работает Illumina NGS?
В технологии Illumina NGS используется подход, принципиально отличающийся от классического метода обрыва цепи по Сэнгеру. Он использует технологию секвенирования путем синтеза (SBS) — отслеживание добавления помеченных нуклеотидов по мере копирования цепочки ДНК — массово-параллельно.
Секвенирование следующего поколения позволяет получить массу данных секвенирования ДНК, оно дешевле и требует меньше времени, чем традиционное секвенирование по Сэнгеру. 2 Системы секвенирования Illumina могут выдавать данные в диапазоне от 300 килобаз до нескольких терабаз за один прогон, в зависимости от типа прибора и конфигурации.
Узнайте больше о технологии SBS
Ключевые преимущества NGS
Доступное секвенирование всего генома
С помощью секвенирования по Сэнгеру на основе капиллярного электрофореза проект «Геном человека» занял более 10 лет и стоил почти 3 миллиарда долларов.
Секвенирование нового поколения, напротив, делает крупномасштабное полногеномное секвенирование (WGS) доступным и практичным для среднего исследователя. Это позволяет ученым анализировать весь геном человека в одном эксперименте по секвенированию или секвенировать от тысяч до десятков тысяч геномов за один год.
Узнайте больше о WGS
Широкий динамический диапазон для профилирования экспрессии
RNA-Seq на основе NGS — это мощный метод, который позволяет исследователям преодолевать неэффективность и дороговизну устаревших технологий, таких как микрочипы. Измерение экспрессии генов микрочипов ограничено шумом на нижнем уровне и насыщением сигнала на верхнем уровне.
В отличие от этого, секвенирование следующего поколения позволяет количественно измерить дискретное цифровое секвенирование, предлагая более широкий динамический диапазон. 1,2,3
Сравните массивы и РНК-Seq
Настраиваемое разрешение для целевых NGS
Целевое секвенирование позволяет вам секвенировать подмножество генов или определенные интересующие области генома, эффективно и экономично фокусируя мощность НГС. NGS обладает высокой масштабируемостью, что позволяет настраивать уровень разрешения в соответствии с экспериментальными потребностями. Выберите, следует ли выполнять поверхностное сканирование нескольких образцов или выполнять секвенирование на большей глубине с меньшим количеством образцов, чтобы найти редкие варианты в заданной области.
NGS обладает высокой масштабируемостью, что позволяет настраивать уровень разрешения в соответствии с экспериментальными потребностями. Выберите, следует ли выполнять поверхностное сканирование нескольких образцов или выполнять секвенирование на большей глубине с меньшим количеством образцов, чтобы найти редкие варианты в заданной области.
Узнайте больше о:
- Целевое секвенирование
- NGS против секвенирования по Сэнгеру
- NGS против количественной ПЦР
Достижения в технологии NGS
Последние достижения компании Illumina в области секвенирования следующего поколения включают:
- Полупроводниковое секвенирование: система iSeq 100 сочетает в себе комплементарный металлооксидный полупроводниковый (CMOS) чип с одноканальным SBS для получения высокоточных данных в компактной системе. Технология проточной кюветы
- : эта опция обеспечивает исключительный уровень производительности для различных приложений секвенирования.
- 75 передовых инноваций: Системы NextSeq 1000 и 2000 обеспечивают гибкость для новых приложений, простой рабочий процесс и анализ данных всего за 2 часа.
- До 16 ТБ: серия NovaSeq X обеспечивает исключительную мощность секвенирования для приложений, интенсивно использующих данные.

Точность Здоровье
Индивидуальные медицинские программы могут помочь пациентам подобрать лечение на основе их генетических чертежей и повысить показатели выживаемости, качество жизни и стоимость лечения.
ПодробнееКак ученые используют NGS
Узнайте, как исследователи в различных областях используют секвенирование нового поколения для совершения прорывных открытий.
Генетика восприимчивости к COVID-19
Это общебританское исследование использует NGS для сравнения геномов тяжелобольных и легкобольных COVID-19пациентов, чтобы помочь раскрыть генетические факторы, связанные с восприимчивостью.
Прочитать статью
Изучение микроокружения опухоли
Ученые используют методы NGS для изучения микроокружения рака, выяснения паттернов экспрессии генов и получения информации о лекарственной устойчивости и метастазировании.
Прочитать статью
Внеклеточная РНК как неинвазивный биомаркер
Это исследование подчеркивает широкий потенциал секвенирования циркулирующей внеклеточной РНК для обнаружения биомаркеров и неинвазивного мониторинга состояния здоровья.
Прочитать статью
˝
Начать использовать NGS
Руководство по покупке системы
Приведенные ниже ресурсы содержат ценные рекомендации для ученых, которые рассматривают возможность приобретения системы секвенирования следующего поколения.
- Просмотр таблиц сравнения платформ NGS
- Воспользуйтесь нашим интерактивным инструментом выбора платформы
- Ознакомьтесь с нашим Руководством покупателя системы NGS, чтобы определить, какие факторы следует учитывать перед покупкой.
Экспериментальные соображения NGS
Узнайте о длине считывания, охвате, показателях качества и других экспериментальных соображениях, которые помогут вам спланировать цикл секвенирования.
Используйте наши интерактивные инструменты, чтобы помочь вам создать собственный протокол NGS или выбрать правильные продукты и методы для вашего проекта.
Начало планирования экспериментов
Ресурсы для высокопроизводительных лабораторий NGS
- Высокопроизводительное секвенирование: обработка большего количества образцов для повышения статистической достоверности. Экономично запускайте мультиомные приложения с большим объемом данных, используя новейшие крупномасштабные секвенсоры.
- Автоматизация подготовки библиотек: изучите автоматизированные решения для работы с жидкостями, разработанные, чтобы помочь лабораториям подготовить большое количество библиотек NGS.
- Системы управления лабораторной информацией (LIMS): автоматизируйте рабочие процессы, интегрируйте инструменты и управляйте образцами.
- Хранилище данных NGS: Надежно храните огромное количество данных NGS и других геномных данных.
Хотите узнать больше о том, как выполнять NGS? Зарегистрируйтесь, чтобы торговый представитель связался с вами.
Вебинары
Гибкие решения с использованием NextSeq 1000 и 2000
Посмотрите записанное обсуждение того, как секвенсоры NextSeq 1000/2000 обеспечивают гибкость и масштабируемость для работы с разнообразными приложениями и методами для эффективных открытий.
Новое определение NGS в исследованиях рака
Эксперты представляют обзор достижений и проблем NGS в продвижении исследований рака, включая обсуждение того, как интегрированный мультиомный подход может быть использован в будущей диагностике и лечении рака.
Использование возможностей геномики для наблюдения за вариантами SARS-CoV-2
Узнайте о методах наблюдения за SARS-CoV-2, включая требования, рабочий процесс и анализ.
Новости геномики
Исследование жидкой биопсии дает новую надежду пациентам с раком легких
Правительство Уэльса запускает программу, направленную на ускорение диагностики и улучшение клинических результатов
Прочитать статьюИспользование аналитики для улучшения диагностики рака и выбора терапии
Разработка и автоматизация рабочих процессов для анализа, обработки и обмена геномными данными между исследователями и клиницистами.